| Parallel Bioinformatics | ||||||||||
|
|
Project
Overview
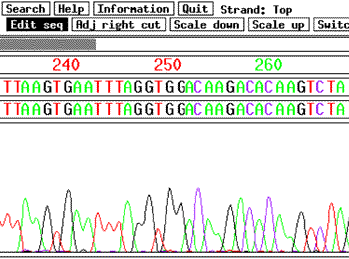
DNA and Protein sequence analysis problems are often both computationally and data intensive. This project focuses on working hand-in-hand with Biologists and Biochemists on the design and implementation of parallel tools for fundamental problems in DNA and Protein sequence analysis and phylogeny. DNA and Protein sequence analysis, to be effective, requires high performance computational tools. Unfortunately, many of the problems involved are NP-complete (i.e. require exponential time unless P=NP). For Biochemists, this computational complexity barrier has created a big problem in that it is often impossible to analyze the much larger new data sets that have recently become available. One possible approach is the use of heuristics. However, these methods introduce unnecessary inaccuracies and, even worse, there is often not even an estimate available for the size of the error introduced by these heuristics. A new approach, which was recently developed by Fellows et.al., is the theory of fixed parameter tractability (FPT). These new FPT algorithms are able to solve NP-complete problem instance that are much larger than what was previously possible, thereby enabling Biochemists to analyze data sets that were previously impossible to process. The goal of my research project is to study how parallelism can further increase the size of problem instances that can be solved via FPT methods. An important example is the vertex cover problem which arises when biochemists analyze protein sequences for large sets of different species, in order to create phylogenetic trees. The vertex cover problem is NP-complete and creates a major limitation on the number of species that biochemists are able to compare. My collaborators and I have designed and implemented a novel parallel FPT method for the vertex cover problem and tested it on a Sun Fire 6800 and PC-cluster. For Biochemists, the only important test for any such new method is how large a problem it can solve. The output size $k$ is the main parameter for the complexity of the vertex cover problem. With sequential FPT methods, the vertex cover problem had previously been considered solvable, within 8 hours, for $k$ up to 200 (Computational Biology group at the ETH Zuerich). In contrast, our parallel code is able to solve instances with $k$=400 in approximately 60 minutes. This is a significant improvement since the time of the sequential FPT algorithm grows exponentially in $k$. It also enables the analysis of certain protein sequence sets (with more than 1000 sequences) which were previously impossible to process. Mike Langston's group at the University of Tennessee has subsequently started to build a parallel FPT method that can be run on a grid platform, thereby further increasing the number of processors available. We have recently combined our efforts with the goal of creating a single, integrated multiprocessor solution which Biochemists can access through a web portal. Our vertex cover code needs to be used in conjunction with CLUSTAL W, a standard software package for sequence alignment. It turns out that our vertex cover code has become so fast that the sequence alignment through Clustal W is becoming a bottleneck. In response, we have designed and implemented a parallel version of CLUSTAL W for PC clusters to become part of our portal. Using our parallel FPT and parallel CLUSTAL code, researchers from Carleton's Biochemistry department have started analyzing Protein sequences for large sets of species that were previously too large to process with conventional methods. The Canadian Bioinformatics Resource has indicated that they will include our portal in their computational ``toolbox'' for Biochemists.
|
|||||||||
|
Software
ClustalXP is our parallel version of Clustal with intergrated support for identifying "bad" sequences is now available for use.
|
||||||||||
| Project
Team
This project represents ongoing joint work with a number of researchers including Jim Cheetham, Frank Dehne, Sylvain Pitre, and Peter Taillon of Carleton University, Faisal Abu-Khzam, Michael A. Langston, and Pushkarak Shanbhag of the University of Tennessee, and Andrew Rau-Chaplin at Dalhousie University.
|
||||||||||
|
|
Publications
| |||||||||